1.Introduction
The documentation describes how to submit jobs to the RaptorX ComplexContact server, as well as retrieve and interpret results. For detailed explanation of the algorithms underlying the server please see the relevant papers listed at http://raptorx2.uchicago.edu/ComplexContact/about/.
2.Job Submission and User Account
- Job submission
-

The "Job Identification" section allows users to provide a job name (default is 'my job') and an email address to be used for notification when the job is done (if you are already logged in this field will be pre-filled). An email is NOT required, but STRONGLY RECOMMEND since it can be used to retrieve the results of all your jobs. The email provided in submission also serves as the username of a user.
-

Jobs can also be submitted using a command-line program curl according to the above format. Note that for curl submission, only one sequence is allowed at a time, and only terms with underline are required. The job URL is returned on screen after successful submission.
- User account and job retrieval
-
We strongly suggest that you provide an email address in submission, which will facilitate the retrieval of your jobs. A new user account identified by your email address is automatically created when you submit your first job. In the case that you do not provide an email address, you may check the status of your job by its server-assigned JobID or its sequence. In case that you cannot find your jobs by all these methods, please contact the RaptorX team.

When your account is first created (after you submitted your first job) you are automatically logged into the server on the machine you are using at that point. If your login session has expired or you want to access your account from a different machine, you may go to the server front page http://raptorx2.uchicago.edu and supply your account email in the login field on the right. Few minutes after submission of the form you will receive an email notification containing a hyperlink to the page containing the jobs for the account.
- Notes
-
A single job may contain at most 50 sequences and each user can have no more than 500 sequences in pending at any time. Further, the results of a job are guaranteed to be stored for only 14 days after the job is completed, although empirically all the current jobs are stored for two years. To store your jobs for a much longer time for publications, please contact the RaptorX team through the "Inquiry & Bug Report" link.
3.Job Monitoring and Job Availability
The "Job Identification" section allows users to provide a job name (default is 'my job') and an email address to be used for notification when the job is done (if you are already logged in this field will be pre-filled). An email is NOT required, but STRONGLY RECOMMEND since it can be used to retrieve the results of all your jobs. The email provided in submission also serves as the username of a user.
Jobs can also be submitted using a command-line program curl according to the above format. Note that for curl submission, only one sequence is allowed at a time, and only terms with underline are required. The job URL is returned on screen after successful submission.
We strongly suggest that you provide an email address in submission, which will facilitate the retrieval of your jobs. A new user account identified by your email address is automatically created when you submit your first job. In the case that you do not provide an email address, you may check the status of your job by its server-assigned JobID or its sequence. In case that you cannot find your jobs by all these methods, please contact the RaptorX team.
When your account is first created (after you submitted your first job) you are automatically logged into the server on the machine you are using at that point. If your login session has expired or you want to access your account from a different machine, you may go to the server front page http://raptorx2.uchicago.edu and supply your account email in the login field on the right. Few minutes after submission of the form you will receive an email notification containing a hyperlink to the page containing the jobs for the account.
A single job may contain at most 50 sequences and each user can have no more than 500 sequences in pending at any time. Further, the results of a job are guaranteed to be stored for only 14 days after the job is completed, although empirically all the current jobs are stored for two years. To store your jobs for a much longer time for publications, please contact the RaptorX team through the "Inquiry & Bug Report" link.
There are three ways to retrieve your jobs: (a) by JobID, (b) by email address, and (c) by sequence.
- By email address
-
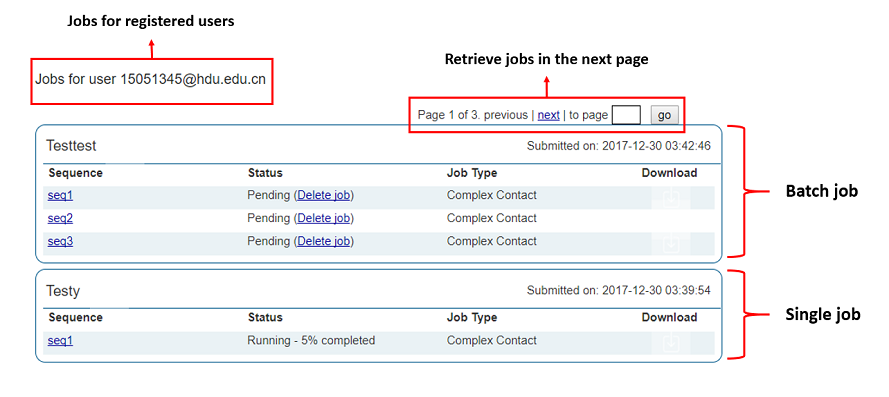
The user needs to be logged in to the server. Refer to Section 2 of this manual for login instructions. Once logged in to the server, selecting "My Jobs" in the menu at the top of the page display a job overview page similar to the one depicted below. Here the status of each prediction in the job is given along with overall information of the predictions being done for each sequence submitted. To track the job status in real-time simply refresh the page and the completion status of the prediction submitted for each sequence in a job will be updated. Clicking on a sequence name will take the user to the result page for this sequence.
- By job ID or sequence
-
Use the "Retrieve Results by JobID" or the "Job status" link. The remaining procedure is similar to job retrieval by email address.

4.Interpretation of Results
We provide three prediction results: (a) a predicted interfacial contact map and (b) a detailed result file.
- Predicted interfacial contact map
-
For the interfacial contact map, we display the probability of two residues being in contact (i.e., their C-beta distance falling in the range [0,8A]). Higher probabilities are represented by darker color.
- Contact result file
-
The contact prediction results in a '.RR' formatted file for each input protein sequence. This file follows the format used in CASP ROLL, click this link for details.

5. Example
Please click the following link to see an example.
6. More Experimental Results
This section presents the experimental results of ComplexContact and its comparison with the others. See our manuscripts for more details.
-
Test results on Baker’s dataset (of 32 protein pairs) and EVComplex’s dataset (of 86 protein pairs)
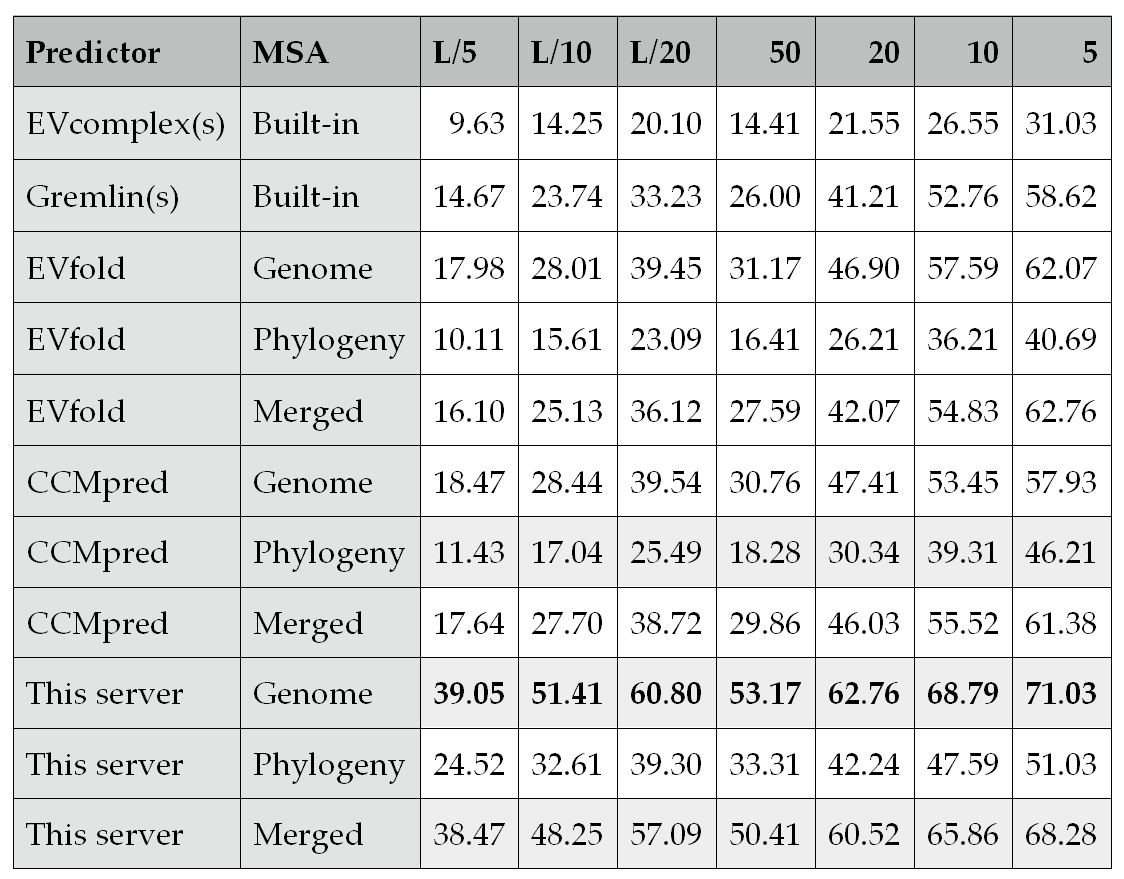
Table 1.Inter-protein contact prediction accuracy (%) on Baker’s data. A method ending with (s) is a web server. EVfold is the same as EVcomplex except that the former uses our MSAs while the latter builds its own MSAs. “Genome” and “Phylogeny” denote genome- and phylogeny-based methods for MSA concatenation, respectively. “Merged” indicates prediction is merged from “Genome” and “Phylogeny”. Columns 3-9 show top-k prediction accuracy for k = L/5, L/10, L/20, 50, 20, 10, and 5, respectively, where L is the length sum of a protein pair.
-
Table 2.Inter-protein contact prediction accuracy (%) on EVComplex’s data. See Table 1 for more explanation.

-
Prediction accuracy on ~ 4500 protein pairs extracted from 3DComplex
The below figure shows the top L/10 inter-protein contact prediction accuracy obtained by this server and CCMpred on the ~4500 protein pairs extracted from 3DComplex using MSAs derived from two different strategies. One dot in the figure represents the prediction accuracy of two methods for one test protein pair. We color a test protein pair by its species. Meanwhile, “N/A” indicates all species other than eukaryotes and prokaryotes. On most test protein pairs, this server outperforms CCMpred by a large margin regardless of their species and how their MSAs are constructed.

-
Correlation between residue-residue distance and predicted probability
From each protein pair in contact, we collect top 100 predicted probability values and examine their correlation with native distance. We discretize residue-residue distance into 6 bins (<4Å, 4-6Å,6-8Å, 8-10Å, 10-12Å and >12Å) and then draw the violin graph to show the distribution of the predicted probability in each doistance bin. As shown in the below figure, the predicted probability decreases along with the distance increases. That is, the smaller the distance is, the higher the predicted probability. Similar to other methods, the distance between two residues is defined as the minimum Euclidean distance of two backbone non-hydrogen atoms.

-
Distribution of predicted probability values for protein pairs in contact vs. protein pairs not in contact
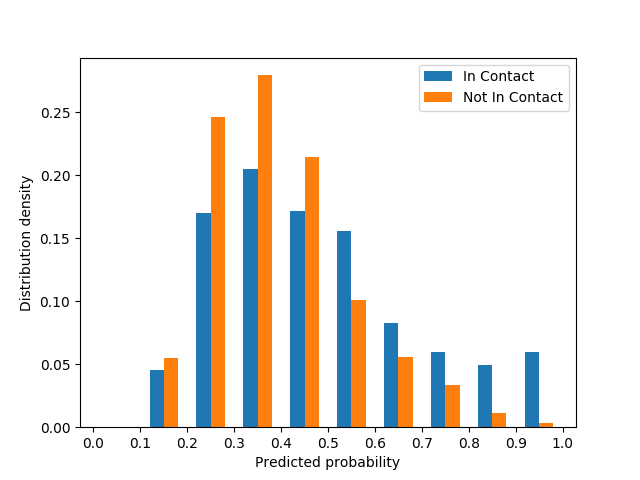
We selected 50 protein pairs with the largest interfacial contact density and ln(meff)≥4 as the positive examples where meff is the number of non-redundant sequences in an MSA and then randomly selected 50 protein pairs (not in contact) as negative control. A pair of proteins are not in contact if they are present in the same protein complex, but their inter-protein residue-residue distances are at least 8Å. From each protein pair, we collect top 50 predicted probability values and then examine the distribution of all probability values. As shown in the below figure, the predicted probability values for a pair of protein chains in contact on average are larger than those for a pair of protein chains not in contact.
-
Distribution of predicted probability values for biological contacts vs. crystal contacts
We have studied 100 protein pairs with the largest contact density and ln(meff)≥4. We generate their crystal complexes by running them on the WHAT IF server (Touw et al., 2015) and then examine the predicted probability value distribution for the biological contacts in a biological complex and the crystal contacts in a crystal complex. Again we say two residues form a native or crystal contact if the minimum Euclidean distance between their backbone non-hydrogen atoms is less than 6Å. As shown in the below figure, on average our DL method generates larger predicted probability value for biological contacts than for crystal contacts.

- Reference
Touw, W. G., Baakman, C., Black, J., te Beek, T. A., Krieger, E., Joosten, R. P., & Vriend, G. (2015). A series of PDB-related databanks for everyday needs. Nucleic Acids Res, 43(Database issue), D364-368. doi:10.1093/nar/gku1028